A Scalable and Quantum‑Accurate Foundation Model for Biomolecular Force Field via Linearly Tensorized Quadrangle Attention
Qun Su†, Kai Zhu†, Qiaolin Gou†, Jintu Zhang, Renling Hu, Yurong Li, Yongze Wang, Hui Zhang, Ziyi You, Linlong Jiang, Yu Kang, Jike Wang, Chang‑Yu Hsieh, Tingjun Hou
arXiv preprint arXiv:2507.00884
PDF
Abstract
arXiv
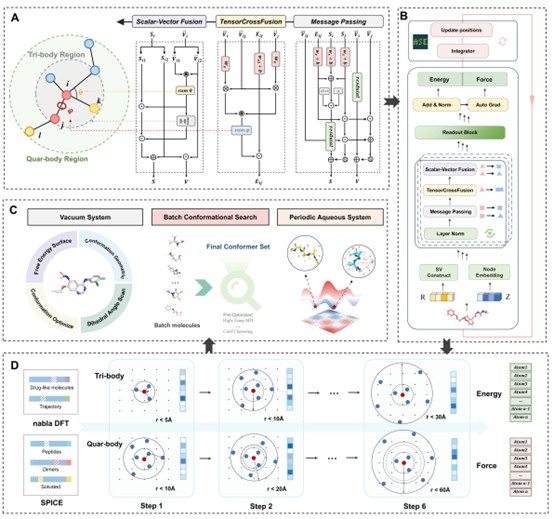
Accurate atomistic biomolecular simulations are vital for disease mechanism understanding, drug discovery, and biomaterial design, but existing simulation methods exhibit significant limitations. Classical force fields are efficient but lack accuracy for transition states and fine conformational details critical in many chemical and biological processes. Quantum Mechanics (QM) methods are highly accurate but computationally infeasible for large-scale or long-time simulations. AI-based force fields (AIFFs) aim to achieve QM-level accuracy with efficiency but struggle to balance many-body modeling complexity, accuracy, and speed, often constrained by limited training data and insufficient validation for generalizability. To overcome these challenges, we introduce LiTEN, a novel equivariant neural network with Tensorized Quadrangle Attention (TQA). TQA efficiently models three- and four-body interactions with linear complexity by reparameterizing high-order tensor features via vector operations, avoiding costly spherical harmonics. Building on LiTEN, LiTEN-FF is a robust AIFF foundation model, pre-trained on the extensive nablaDFT dataset for broad chemical generalization and fine-tuned on SPICE for accurate solvated system simulations. LiTEN achieves state-of-the-art (SOTA) performance across most evaluation subsets of rMD17, MD22, and Chignolin, outperforming leading models such as MACE, NequIP, and EquiFormer. LiTEN-FF enables the most comprehensive suite of downstream biomolecular modeling tasks to date, including QM-level conformer searches, geometry optimization, and free energy surface construction, while offering 10x faster inference than MACE-OFF for large biomolecules (~1000 atoms). In summary, we present a physically grounded, highly efficient framework that advances complex biomolecular modeling, providing a versatile foundation for drug discovery and related applications.